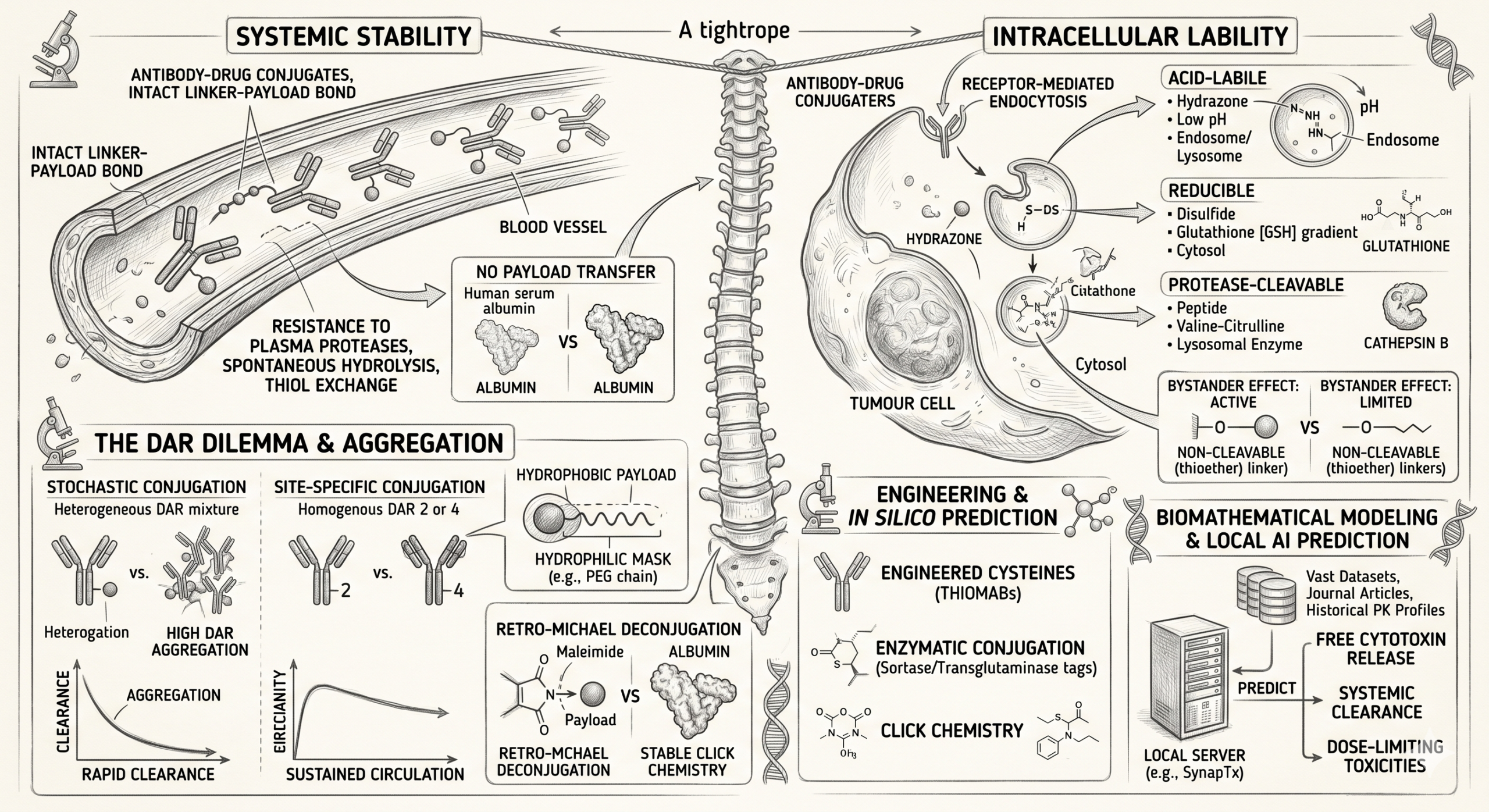
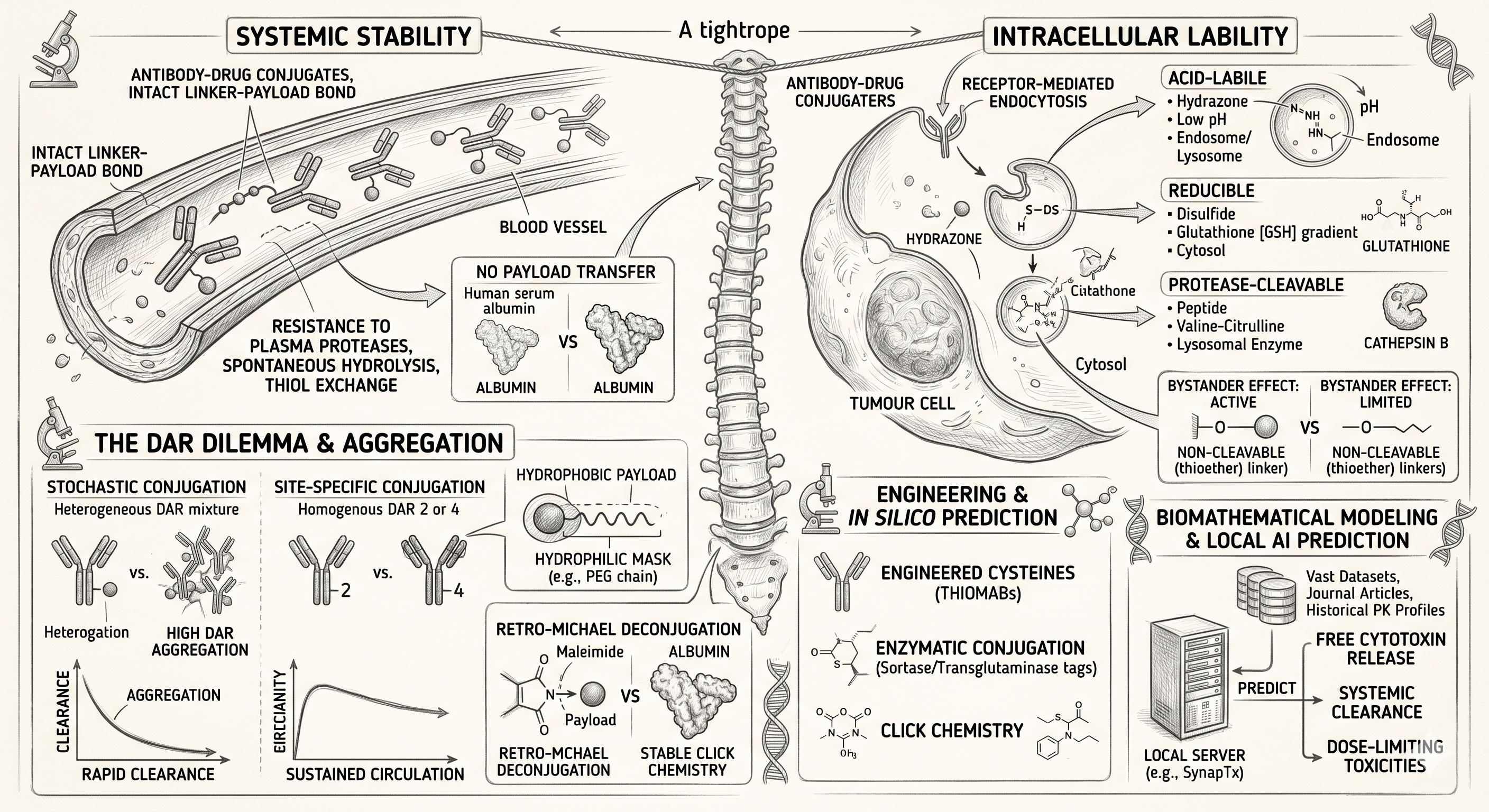
The Crucial Science of ADC Linker Stability
The linker is far more than a chemical bridge — it is the primary determinant of an ADC's pharmacokinetic profile, safety, and ultimate clinical success. If the linker fails, the entire programme fails.
The advent of Antibody-Drug Conjugates (ADCs) has fundamentally reshaped the precision oncology landscape. By tethering highly potent cytotoxic payloads to monoclonal antibodies, ADCs promise a 'magic bullet' approach — delivering catastrophic payloads directly to malignant cells whilst sparing healthy tissue.
However, beneath the elegant conceptual framework of an ADC lies one of the most formidable bioengineering challenges in modern drug development: the linker. This analysis examines the complex dynamics of linker stability, exploring why bridging the gap between systemic inertness and targeted release is the most precarious tightrope in targeted therapeutics.
Systemic Stability
Upon intravenous administration, the ADC must navigate systemic circulation for days or even weeks. The linker must remain completely intact — resisting enzymatic degradation by plasma proteases, spontaneous hydrolysis, and thiol exchange. Premature cleavage releases a naked cytotoxin 100 to 1,000 times more potent than conventional chemotherapy, causing severe dose-limiting off-target toxicities and a drastically narrowed therapeutic index.
Intracellular Lability
Once the ADC binds to its target antigen on the tumour cell surface and undergoes receptor-mediated endocytosis, the linker must rapidly and efficiently self-destruct. Whether triggered by the acidic environment of the endosome, the enzymatic milieu of the lysosome, or the reducing conditions of the cytosol, the linker must snap to release the active payload precisely at the site of action.
"Balancing ultra-stability in the blood and rapid lability within the tumour — this is the crux of modern ADC formulation."
The Chemistry of Deconjugation and Premature Release
To understand linker stability, one must examine the mechanisms of premature payload loss. Early-generation ADCs heavily relied on classical maleimide chemistry for conjugation to the antibody's cysteine residues. Whilst efficient, maleimide linkages are notoriously susceptible to a reverse reaction known as retro-Michael deconjugation.
In systemic circulation, the maleimide-thiol bond can spontaneously break, releasing the linker-payload complex. This freed complex can then undergo a secondary Michael addition, irreversibly binding to reactive thiols on circulating serum albumin — transforming serum albumin into a long-circulating carrier of highly toxic payloads and driving severe systemic adverse events.
Modern engineering has pivoted towards self-hydrolysing maleimides or entirely novel 'click chemistry' approaches to lock these bonds in place and prevent plasma-mediated deconjugation.
The Primary Linker Paradigms: Cleavable vs. Non-Cleavable
The strategies employed to achieve this delicate balance broadly fall into two distinct structural categories, each possessing unique pharmacokinetic advantages and limitations.
Cleavable Linkers: Exploiting the Tumour Microenvironment
Cleavable linkers are designed to exploit the stark physiological differences between systemic circulation and the intracellular or tumour microenvironment (TME).
Acid-Labile Linkers (Hydrazones)
Among the first utilised in the clinic, these remain relatively stable at the neutral pH of blood (pH 7.4) but undergo rapid hydrolysis in the acidic environments of endosomes (pH 5.0–6.0) and lysosomes (pH 4.8). However, clinical data has shown that hydrazones exhibit undesirable slow hydrolysis in systemic circulation over time, contributing to significant off-target toxicity.
Reducible Linkers (Disulfides)
These linkers exploit the dramatic concentration gradient of glutathione — a highly reducing tripeptide. Glutathione concentrations are significantly higher intracellularly (and specifically within tumour cells due to oxidative stress) compared to blood plasma. Once internalised, the disulfide bond is reduced, liberating the payload.
Protease-Cleavable Linkers (Peptides)
Currently the industry gold standard, these linkers — such as the widely used valine-citrulline dipeptide — are exceptionally stable in plasma due to the presence of protease inhibitors in the blood. They are designed to be selectively cleaved by specific lysosomal enzymes such as Cathepsin B, which is heavily overexpressed in numerous malignancies, providing a highly controlled and predictable release profile.
Non-Cleavable Linkers: The Reliance on Catabolism
Non-cleavable linkers, typically relying on robust thioether bonds, take a fundamentally different approach — possessing no specific chemical trigger. Instead, they rely entirely on the comprehensive proteolytic degradation of the monoclonal antibody backbone within the lysosome.
Once the ADC is internalised, the antibody is digested into its constituent amino acids, leaving the cytotoxic payload attached to a small amino acid remnant of the linker. Because the thioether bond is virtually indestructible in systemic circulation, these ADCs boast superior plasma stability and extended half-lives. However, the amino-acid-tethered payload often struggles to cross cell membranes, effectively eliminating the bystander effect — the ability of a released drug to diffuse into and kill adjacent, target-negative tumour cells.
The DAR Dilemma and the Threat of Aggregation
When discussing linker stability, one cannot ignore its direct impact on the Drug-to-Antibody Ratio (DAR) — the average number of payload molecules attached to each antibody. Historically, early-generation ADCs utilised stochastic (random) conjugation methods, attaching payloads to native lysines or reduced cysteines. This yielded a highly heterogeneous mixture of species, ranging from naked antibodies (DAR 0) to heavily loaded molecules (DAR 8 or higher).
To combat hydrophobicity-induced instability, modern linker design frequently incorporates hydrophilic masks such as discrete polyethylene glycol (PEG) chains. By PEGylating the linker architecture, formulators can shield the hydrophobic payload — stabilising the entire conjugate in plasma, preventing aggregation, and drastically extending the circulatory half-life.
Engineering Stability: The Shift to Site-Specific Conjugation
To overcome the instability inherent in stochastic conjugation and retro-Michael reactions, the industry has aggressively pivoted towards site-specific conjugation. By precisely controlling where the linker attaches to the antibody, developers can place the payload in microenvironments that naturally shield the chemical bond from systemic hydrolysis.
Engineered Cysteines (THIOMABs)
By substituting specific amino acids on the antibody backbone with reactive cysteines, scientists can direct the linker to attach at sterically hindered sites. This spatial shielding protects the linker-payload bond from circulating reducing agents in the blood, vastly improving systemic stability whilst preserving intracellular lability.
Enzymatic Conjugation
Technologies utilising enzymes such as microbial transglutaminase or sortase allow for the creation of robust, unnatural peptide bonds between the linker and specific glutamine or glycine tags engineered into the antibody. These bonds exhibit exceptional stability in plasma.
Click Chemistry
The application of bio-orthogonal 'click chemistry' — most notably strain-promoted azide-alkyne cycloaddition (SPAAC) — has revolutionised linker attachment. These reactions form incredibly stable triazole rings that are entirely inert to the native biological environment, effectively eliminating the risk of premature deconjugation in the bloodstream.
Biostatistics and In Silico Prediction: The New Frontier
The biological complexity of human plasma and the tumour microenvironment means that empirical in vitro testing of linker stability often fails to scale accurately to human trials. Because a linker's degradation rate dictates the systemic release of the free cytotoxin, accurately modelling its pharmacokinetic profile is paramount to predicting dose-limiting toxicities.
To bridge this translational gap, modern drug development programmes are increasingly relying on advanced biostatistical frameworks and deep in silico predictive modelling.
By deploying robust, localised AI architectures to process vast datasets of molecular descriptors and historical PK profiles, biostatisticians can map the predicted systemic clearance and enzymatic cleavage rates of novel linker designs before they ever enter a physical laboratory. These highly parameterised models allow teams to simulate how a specific linker architecture will behave across varying human metabolic profiles — identifying potential liabilities such as unexpected plasma protease cleavage and enabling the rational design of safer, more stable ADCs.
The evolution of the Antibody-Drug Conjugate is fundamentally the evolution of the linker. We have transitioned from viewing the linker as a simple chemical tether to understanding it as a highly sophisticated, conditionally active machine.
As we refine site-specific conjugation methodologies, integrate hydrophilic shielding, and leverage advanced computational biostatistics to predict human pharmacokinetics, the therapeutic window of ADCs will continue to widen. The ultimate goal of precision oncology — maximum lethality at the tumour site with zero systemic collateral damage — relies entirely on mastering this microscopic tightrope of stability.